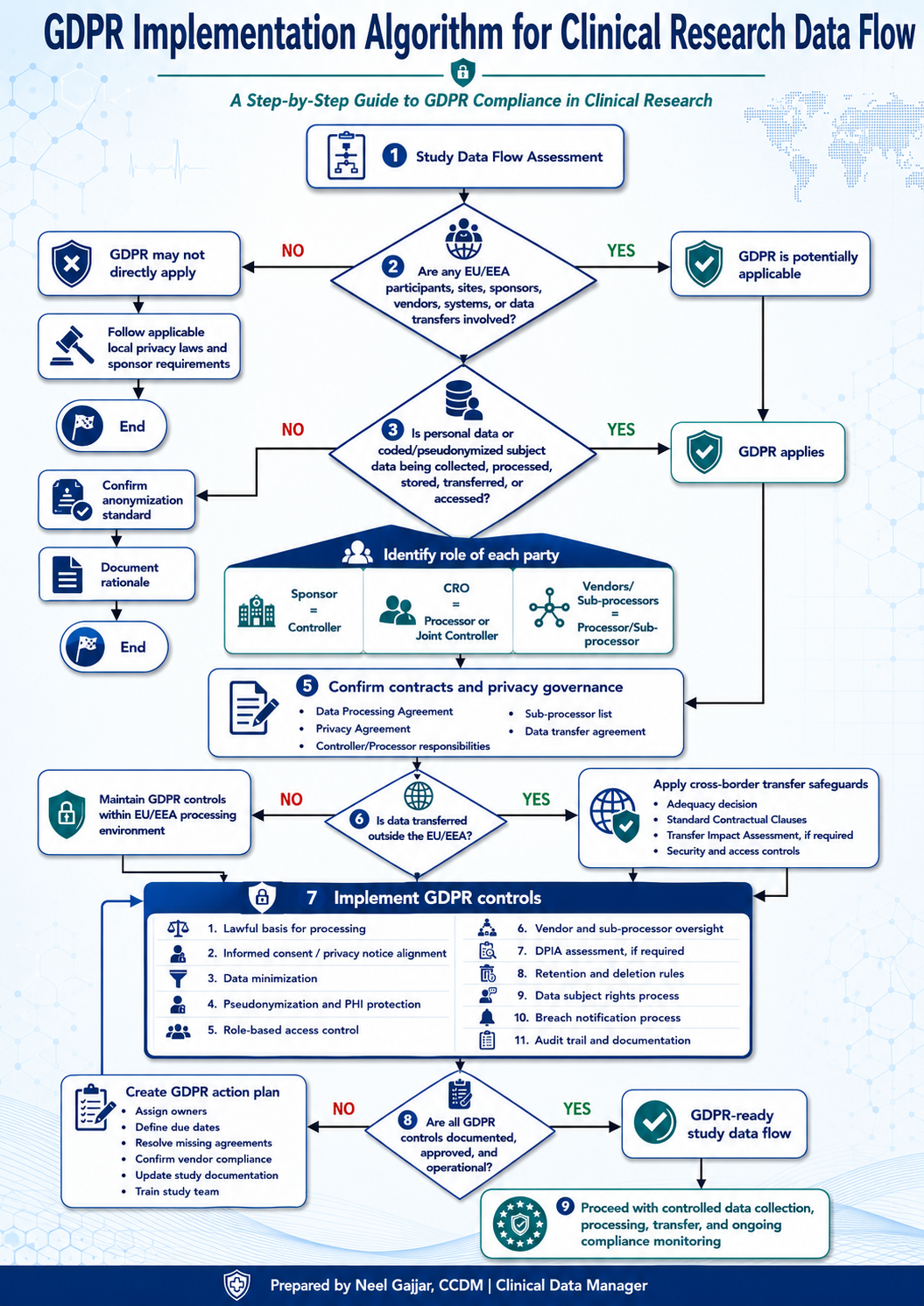
GDPR Implementation Algorithm for Clinical Study Data Flow
A Practical Study Start-Up Framework for CROs, Sponsors, and Clinical Data Management Teams
A study team is preparing for EDC go-live.
The sponsor is located in the EU. Sites may be located in multiple countries. The EDC is hosted by a vendor. The CRO will export data into a controlled folder. A lab vendor will send external data files. The sponsor DM team will receive interim and final datasets. The PM asks:
“Are we GDPR-ready?”
This is where many study teams struggle.
GDPR is often treated as a legal or consent-form topic, but in clinical research it is also an operational data governance issue. It affects how data is collected, where data is hosted, who can access it, how vendors are approved, how exports are controlled, how privacy language is written, how UAT is performed, and what evidence is filed before go-live.
For Clinical Data Management teams, GDPR should not be handled only after the database is built. It should be considered during protocol review, eCRF design, vendor setup, DMP development, access planning, UAT, data transfer setup, and TMF/evidence filing.
This article presents a practical GDPR Implementation Algorithm for Clinical Study Data Flow that can be used during study start-up before EDC build or go-live.
The Problem This Framework Solves
Modern clinical trial data does not stay in one place.
It may move through:
Study sites
Source documents
EDC systems
ePRO/eCOA platforms
Laboratory vendors
Device or wearable vendors
Imaging or ECG vendors
Safety/PV systems
CRO-controlled folders
Sponsor data teams
Statistical programmers
Cloud storage systems
Regulatory archives
Because of this, the main challenge is not simply asking, “Does GDPR apply?”
The better question is:
“How do we operationally implement GDPR across the complete study data flow and prove that the controls were in place before go-live?”
The algorithm helps study teams manage:
GDPR applicability
GDPR trigger source
Controller/processor role mapping
DPA and contract status
Special-category data and legal basis
Vendor/sub-processor oversight
International transfer assessment
Data minimization
Privacy-by-design in EDC
ICF/privacy notice alignment
DPIA screening
Privacy UAT
Data subject rights handling
Breach escalation
Evidence filing
Go-live readiness
Simple GDPR Glossary for Clinical Research Teams
Term | Simple Meaning in a Clinical Study |
Personal data | Any information that can identify a person directly or indirectly |
Coded/pseudonymized data | Data where direct identifiers are replaced with a code, but re-identification may still be possible |
Controller | Party that decides why and how personal data is processed; often the sponsor |
Processor | Party that processes data on behalf of the controller; often the CRO or vendor |
Sub-processor | A vendor used by a processor to process study data |
DPA | Data Processing Agreement defining processor obligations |
DPIA | Data Protection Impact Assessment for high-risk processing |
Special-category data | Sensitive data such as health, genetic, biometric, or similar data |
Transfer safeguard | GDPR mechanism or control used when data moves internationally |
Privacy-by-design | Building privacy controls into study systems and processes from the beginning |
One important point for CDM teams: coded clinical trial data may still be personal data under GDPR if someone can reasonably link it back to a participant using a key or other information.
Generic Clinical Study Data Flow
The algorithm starts by documenting the actual study data flow.
A generic clinical study data flow can be written as:
Study Site / Source Data → EDC or eClinical System → CRO Processing / Storage Location → Sponsor / Recipient Location
This model is intentionally flexible. It can apply to any study, regardless of the country, CRO, sponsor, EDC system, or vendor setup.
Before deciding whether GDPR applies, the team should document:
Data Flow Element | Key Question |
Participant location | Are any participants located in the EU/EEA? |
Site location | Are any clinical sites located in the EU/EEA? |
Sponsor location | Is the sponsor/controller located in the EU/EEA? |
CRO location | Is the CRO/processor located in the EU/EEA? |
EDC system | Which system is used for data capture? |
EDC hosting location | Where is the EDC hosted? |
CRO storage location | Where are exports and listings stored? |
Vendor locations | Which vendors process data and where are they located? |
Support access location | Can vendor support teams access data from another country? |
Transfer route | From which country to which country does data move? |
The output should be a study data flow diagram and a hosting/transfer summary.
GDPR Implementation Algorithm
Step 1: Confirm GDPR Applicability
The first decision is whether GDPR applies to the study.
Ask:
Are any participants located in the EU/EEA?
Are any clinical sites located in the EU/EEA?
Is the sponsor or controller located in the EU/EEA?
Is the CRO or processor located in the EU/EEA?
Is the EDC, vendor, or data host located in the EU/EEA?
Will data be transferred from the EU/EEA to a non-EU/EEA country?
Will data be transferred from a non-EU/EEA country to an EU/EEA sponsor/controller?
Does the sponsor contract require GDPR compliance?
Answer | Action |
Yes | Proceed with GDPR implementation steps |
No | Document rationale why GDPR does not apply |
Not sure | Escalate to Sponsor Legal/Privacy before EDC build or go-live |
Key output: GDPR Applicability Assessment
Step 2: Identify the GDPR Trigger Source
It is not enough to say GDPR applies. The study team should document why GDPR applies.
Possible trigger sources include:
EU/EEA participant involvement
EU/EEA site involvement
EU/EEA sponsor/controller involvement
EU/EEA CRO/processor involvement
EU/EEA vendor or data host involvement
EU/EEA data export
EU/EEA data import
Contractual GDPR requirement
This helps explain the privacy decision during sponsor review, QA review, audit, or inspection.
Key output: GDPR Trigger Source Record
Step 3: Map GDPR Roles and Responsibilities
The study team must confirm who is acting as:
Controller
Processor
Joint Controller
Sub-processor
In many studies, the sponsor is the controller and the CRO is the processor. However, this should not be assumed. Sites, vendors, and cloud providers may have different roles depending on the contract and data flow.
Role mapping should include:
Party | Role to Confirm |
Sponsor | Controller / Joint Controller |
CRO | Processor / Controller / Joint Controller |
Clinical site | Controller / Joint Controller / Processor |
EDC vendor | Processor / Sub-processor |
Lab vendor | Processor / Sub-processor |
ePRO/eCOA vendor | Processor / Sub-processor |
Device/wearable vendor | Processor / Sub-processor |
Imaging/ECG vendor | Processor / Sub-processor |
Safety/PV vendor | Processor / Sub-processor |
Cloud/file storage provider | Processor / Sub-processor |
Statistical programming vendor | Processor / Sub-processor |
If roles are confirmed, document them in a Privacy RACI.
If roles are unclear, escalate to Sponsor/CRO Legal before vendor activation or data transfer.
Key output: GDPR Role Map / Privacy RACI
Step 4: Confirm DPA and GDPR Contract Terms
A Data Processing Agreement, or DPA, is required when a processor processes personal data on behalf of a controller.
For a clinical study, the DPA or privacy agreement should define:
· Subject matter and duration of processing
· Nature and purpose of processing
· Types of personal data
· Categories of data subjects
· Controller instructions
· Processor obligations
· Confidentiality
· Security obligations
· Sub-processor rules
· Breach notification timelines
· Audit and inspection support
· Return, deletion, retention, or archival expectations
DPA Status | Action |
DPA in place | File copy or contract reference |
DPA missing | Initiate DPA or privacy addendum |
Not sure | Ask Contracts/Legal to confirm MSA/SOW/DPA status |
Key output: DPA Status Confirmation
Step 5: Confirm Special-Category Data and Legal Basis
Clinical studies commonly involve health data, which is special-category data under GDPR.
The team should confirm whether the study processes:
· Health data
· Genetic data
· Biometric data
· Pregnancy data
· Medical history
· Adverse events
· Concomitant medications
· Laboratory results
· Vital signs
· Procedures
· Safety data
Sponsor Legal/Privacy should confirm:
GDPR Item | Meaning |
Article 6 lawful basis | Why personal data can be processed |
Article 9 condition | Why health/special-category data can be processed |
Research safeguards | What protections apply for scientific research |
Consent clarification | Whether clinical trial consent and GDPR legal basis are separate |
The ICF/privacy notice should not be finalized until the legal basis and special-category condition are understood.
Key output: Legal Basis and Article 9 Confirmation
Step 6: Review Vendors and Sub-processors
Every vendor that processes study data should be identified and assessed.
For each vendor/system, document:
Vendor Detail | Why It Matters |
Vendor name | Identifies who processes data |
Service provided | Explains why vendor is used |
GDPR role | Confirms processor/sub-processor status |
Data processed | Defines risk and scope |
Hosting location | Supports transfer assessment |
Support/access location | Identifies remote access risk |
Sub-processors | Shows onward processing |
DPA status | Confirms contractual control |
Sponsor approval | Shows controller oversight |
Security review | Shows technical/organizational safeguards |
This applies to EDC, ePRO/eCOA, lab, device, imaging, ECG, safety, cloud storage, and programming vendors.
Key outputs: Vendor/Sub-processor List, Vendor Privacy/Security Review, Hosting Location Summary
Step 7: Assess Data Flow and International Transfers
The team should assess whether personal data moves into, within, or outside the EU/EEA.
Ask:
· Does data originate in the EU/EEA?
· Does data leave the EU/EEA?
· Does data enter the EU/EEA?
· Is the sponsor located in the EU/EEA?
· Is the CRO outside the EU/EEA?
· Is the EDC/vendor hosted outside the EU/EEA?
· Are vendor support teams accessing data from outside the EU/EEA?
· Are onward transfers involved?
· Are transfer safeguards required?
Use this simple transfer scenario table:
Transfer Scenario | Practical Action |
EU/EEA to EU/EEA | Confirm secure transfer and DPA/role controls |
EU/EEA to adequate country | Confirm adequacy and contract terms |
EU/EEA to non-adequate country | Assess SCCs, TIA, supplementary measures |
Non-EU/EEA to EU sponsor/controller | Confirm sponsor GDPR requirements and processing role |
Onward transfer to another vendor/country | Confirm sub-processor approval and transfer controls |
Operational controls should include:
· Approved transfer method
· Encryption in transit
· Encryption at rest where applicable
· Access restrictions
· Transfer QC
· File/record count checks
· Transfer confirmation
Key outputs: Data Flow Diagram, International Transfer Assessment, Transfer Safeguard Decision, Transfer QC Log
Step 8: Apply Data Minimization and Privacy-by-Design
GDPR should be built into the study design.
From a CDM and EDC perspective, privacy-by-design means:
· Use coded Subject IDs where possible
· Avoid unnecessary direct identifiers
· Minimize free-text fields
· Restrict or redact attachments
· Avoid unnecessary source document uploads
· Control query response content
· Limit reports and exports to necessary data
· Apply role-based and least-privilege access
· Define blinding/unblinding controls if applicable
Even if direct identifiers are not collected, privacy risk can still occur through:
· Free-text fields
· Query responses
· AE/SAE narratives
· Attachments
· External data files
· Reports
· Listings
· Exports
· Audit trail metadata
Add clear language to the CRF Completion Guidelines and site training:
Do not enter participant name, address, phone number, email, health card number, medical record number, or other direct identifiers into EDC free-text fields, query responses, or attachments unless specifically required and approved.
Key outputs: EDC Privacy-by-Design Checklist, Data Minimization Review, Site Training Privacy Instruction
Step 9: Align the ICF and Privacy Notice
The ICF/privacy notice must match the actual study data flow.
It should explain:
· Sponsor/controller identity
· CRO/vendor roles
· Data collected
· Purpose of processing
· Legal basis or special-category condition where appropriate
· International transfers
· Data recipients
· Retention period
· Participant/data subject rights
· Privacy or DPO contact
· Limits on withdrawal/deletion where applicable
A common risk is that the actual data flow changes, but the ICF/privacy notice remains generic or outdated.
For example, if data moves from site to EDC, then to CRO, then to sponsor, and then to a programming vendor, the privacy notice should not imply that only the site and sponsor will access the data.
Key output: ICF / Privacy Notice Review Confirmation
Step 10: Complete DPIA or Privacy Risk Screening
A DPIA may be required when processing is likely to result in high risk to individuals.
Clinical studies should screen for:
· Health data
· Genetic data
· Biometric data
· Vulnerable participants or minors
· Large-scale processing
· Wearable or device data
· Imaging or ECG data
· AI, automation, or new technology
· International transfers
· Direct identifiers
· Multiple vendors or sub-processors
DPIA Decision | Action |
Required | Complete DPIA before go-live or high-risk processing |
Not required | Document rationale |
Not sure | Complete DPIA screening and obtain Sponsor Legal/Privacy decision |
Key outputs: DPIA Report, DPIA Screening Record, Privacy Risk Mitigation Actions
Step 11: Build GDPR Controls into EDC
The EDC should be privacy-ready before go-live.
Key controls include:
· Coded Subject ID
· No unnecessary direct identifiers
· Limited free text
· Attachment or redaction controls
· Role-based access
· Least-privilege permissions
· Export permission controls
· Report access controls
· Audit trail enabled
· Audit trail export/review capability
· Query text guidance
· Site privacy training language
· Blinding/unblinding access controls where applicable
The database should not only be functionally ready. It should also be privacy-ready.
Key outputs: EDC Privacy Control Checklist, eCRF Privacy Review Evidence, Access Role Matrix, Export/Report Permission Review
Step 12: Test Privacy Controls During UAT
Privacy controls should be tested during UAT.
Sample privacy UAT cases:
UAT Test | Expected Result |
Create a subject | System allows coded Subject ID only |
Review demographic fields | No unnecessary name, address, phone, email, MRN, or health card fields |
Site user attempts full export | Export is blocked unless role permits |
DM user performs export | Export includes only approved/coded fields |
Sponsor reviewer accesses reports | Access matches approved role |
Free-text field review | CCG/training instructs not to enter identifiers |
Attachment upload | Disabled or redaction guidance/control is present |
Audit trail test | Change is captured with user, date, time, old value, and new value |
Query response test | Query guidance avoids unnecessary identifiers |
Blinded role access | Blinded user cannot access unblinded data |
If privacy UAT passes, retain evidence.
If privacy UAT fails, resolve defects and retest.
If privacy UAT was not planned, add it before go-live.
Key outputs: Privacy UAT Evidence, Audit Trail Evidence, Access Review Evidence
Step 13: Define Data Subject Rights Handling
The study team should confirm how participant/data subject rights requests will be handled.
Requests may include:
· Access
· Correction
· Restriction
· Objection
· Erasure/deletion
· Withdrawal-related privacy questions
· Portability, if applicable
In many CRO models, the CRO should not independently respond unless authorized. The CRO usually routes requests to the sponsor/controller and supports the response as instructed.
Create a simple rights handling plan:
Request Type | Primary Owner | CRO Role | Evidence |
Access | Sponsor/controller | Support as instructed | Request log |
Correction | Sponsor/site/controller | Support data correction workflow | Query/audit trail |
Restriction | Sponsor/controller | Escalate | Decision record |
Objection | Sponsor/controller | Escalate | Decision record |
Erasure/deletion | Sponsor/controller/legal | Escalate; assess regulatory limits | Decision record |
Withdrawal-related question | Site/sponsor/controller | Support as instructed | Request log |
Key outputs: Data Subject Rights Handling Plan, Escalation Pathway, Request Log Location
Step 14: Define Breach and Privacy Incident Escalation
A GDPR implementation plan should define what happens if a privacy incident occurs.
Confirm:
· Internal CRO escalation route
· Sponsor notification pathway
· Vendor breach notification timeline
· Who decides regulator/participant notification
· Incident log location
· QA/CAPA linkage for significant or systemic incidents
· Evidence retention
This should be defined before go-live because privacy incidents require fast escalation.
Key output: Breach / Privacy Incident Escalation Plan
Step 15: File GDPR Evidence and Prepare for Inspection Readiness
GDPR evidence should be filed in an agreed location such as:
· eTMF
· Sponsor portal
· CRO QMS folder
· Contracts folder
· Vendor oversight folder
· DM study folder
The evidence package should include:
· GDPR Applicability Assessment
· GDPR Trigger Source Record
· GDPR Role Map / Privacy RACI
· DPA Status Confirmation
· Legal Basis / Article 9 Confirmation
· Vendor/Sub-processor List
· Vendor Privacy/Security Review
· Data Flow Diagram
· International Transfer Assessment
· Transfer QC Plan / Log
· ICF / Privacy Notice Review
· DPIA or DPIA Screening Record
· EDC Privacy Control Checklist
· Privacy UAT Evidence
· Access Role Matrix
· Audit Trail Evidence
· Export/Report Permission Review
· Data Subject Rights Handling Plan
· Breach / Privacy Incident Escalation Plan
· Archive / Retention Confirmation
Key output: GDPR Evidence Tracker
Step 16: Apply a Minimum GDPR Go-Live Gate
Before EDC go-live, the study should pass a minimum GDPR go-live gate.
GDPR Go-Live Item | Complete |
Study data flow documented | ☐ |
GDPR applicability confirmed | ☐ |
GDPR trigger source documented | ☐ |
GDPR role mapping completed | ☐ |
DPA/privacy agreement status confirmed | ☐ |
Legal basis and Article 9 condition confirmed | ☐ |
Vendor/sub-processor list completed | ☐ |
Vendor hosting/support locations documented | ☐ |
Data flow diagram approved | ☐ |
Transfer method approved | ☐ |
Transfer safeguard decision documented | ☐ |
Transfer QC plan defined | ☐ |
Data minimization review completed | ☐ |
ICF/privacy notice reviewed | ☐ |
DPIA decision documented | ☐ |
EDC privacy controls implemented | ☐ |
Privacy UAT passed | ☐ |
Access role matrix approved | ☐ |
Audit trail/export controls verified | ☐ |
Data subject rights handling pathway defined | ☐ |
Breach/privacy incident escalation pathway defined | ☐ |
GDPR evidence filing owner/location confirmed | ☐ |
Status | Decision |
All critical items complete | Proceed to EDC go-live |
Minor items open | Proceed only if risk accepted with owner and due date |
Major items open | Do not proceed; escalate to PM, Sponsor Legal/Privacy, CRO QA, and Lead DM |
Step 17: Maintain and Monitor During Study Conduct
GDPR implementation does not end at go-live.
During study conduct, maintain oversight through:
· Periodic access reviews
· Vendor oversight
· Transfer QC
· Privacy issue review
· DPIA updates when data flow changes
· ICF/privacy notice review after relevant amendments
· Data subject rights support
· Breach/privacy incident escalation
· Evidence filing QC
· Archive and retention confirmation
Final output: Ongoing GDPR Oversight Evidence and Closeout GDPR Evidence Package
Example Case Study
Scenario
A clinical study includes:
· Sponsor located in France
· CRO located in the United States
· Sites in Germany and Canada
· EDC hosted in the United States
· ePRO vendor located in Ireland
· Central lab located in Germany
· Statistical programming vendor located in India
How the Algorithm Applies
Algorithm Step | Example Decision |
GDPR applicability | GDPR applies due to EU sponsor, EU sites, EU participants, EU lab, and EU ePRO vendor |
Trigger source | EU sponsor/controller, EU site/participants, EU vendor, international transfers |
Role mapping | Sponsor likely controller; CRO likely processor; vendors likely processors/sub-processors |
DPA | Sponsor-CRO DPA required; vendor DPAs/sub-processor terms required |
Special-category data | Health data and lab data involved; Article 9 condition required |
Vendor review | EDC, ePRO, lab, and programming vendor reviewed |
Transfer assessment | EU/EEA data transferred to US and India; safeguards assessed |
EDC privacy controls | Coded Subject ID, role-based access, audit trail, export limits |
ICF/privacy notice | Must describe international transfers and vendor access |
DPIA screening | Required due to health data, EU data, international transfers, multiple vendors |
UAT | Privacy UAT includes access, exports, audit trail, coded ID |
Evidence | Filed before go-live in agreed study privacy/evidence folder |
This example shows why GDPR cannot be handled only by reviewing the consent form. It must be built into the full study data workflow.
Common Mistakes to Avoid
Common Mistake | Why It Creates Risk |
Assuming coded data is not personal data | Coded/pseudonymized data may still be personal data |
Starting EDC build before confirming data flow | Privacy controls may be missed |
Reviewing ICF before vendor/data flow is known | Privacy language may not match actual processing |
Forgetting vendor hosting/support locations | Remote support access may create cross-border processing |
Not checking sub-processors | Sponsor/controller approval may be missing |
Treating DPA as only a legal issue | DPA affects operations, breach timelines, vendor use, and archival |
Not testing export permissions in UAT | Unauthorized users may export data |
Allowing free-text without guidance | Sites may enter unnecessary identifiers |
Not documenting DPIA rationale | Privacy risk decision may not be inspection-ready |
Not assigning evidence filing owner | Documents may exist but not be filed or QC’d |
Who Owns What?
Activity | Primary Owner | Support |
GDPR applicability decision | Sponsor Legal/Privacy | PM, Lead DM |
Data flow diagram | Lead DM / PM | IT, vendors, Sponsor DM |
GDPR role mapping | Sponsor Legal/Privacy | CRO Legal, PM |
DPA confirmation | Contracts/Legal | PM |
Vendor/sub-processor list | PM / Vendor Manager | Lead DM, IT |
Hosting/support location confirmation | PM / Vendor Manager | IT, vendor |
Transfer assessment | Sponsor Legal/Privacy | PM, Lead DM, IT |
EDC privacy controls | Lead DM / EDC build team | QA, Sponsor |
Privacy UAT | Lead DM / EDC team | QA |
ICF/privacy notice review | Sponsor Legal/Privacy | PM, Clinical, Lead DM |
DPIA decision | Sponsor Privacy/DPO | PM, Lead DM |
Data subject rights handling | Sponsor/controller | CRO supports as instructed |
Breach escalation plan | Sponsor/CRO QA/Privacy | PM, vendors |
GDPR evidence filing | PM / TMF Owner | Lead DM, QA |
How to Use the Flowchart and Checklist Together
The GDPR flowchart should be used as the visual decision guide during study start-up.
The go-live checklist should be used as the operational evidence tracker.
Together, they help the study team answer:
· What decisions are needed?
· Who owns each decision?
· What evidence is required?
· What must be completed before EDC go-live?
· What should be monitored during study conduct?
This makes GDPR implementation practical, trackable, and inspection-ready.
Final Takeaway
GDPR implementation in clinical research is not a one-time legal checkbox.
It should be embedded into:
· Study start-up
· Data flow mapping
· Vendor oversight
· EDC design
· DMP and CCG development
· ICF/privacy notice review
· Data transfer setup
· Access control
· UAT
· Data subject rights handling
· Breach escalation
· Evidence filing
· Closeout and archival
For Clinical Data Management teams, the practical goal is simple:
Collect only what is needed.
Use coded data where possible.
Control access.
Limit free text.
Secure transfers.
Test privacy controls.
Document decisions.
File evidence.
Monitor continuously.
That is how GDPR becomes operational, inspection-ready, and aligned with strong clinical data governance.
About the Author
Neel Gajjar
CCDM®Clinical Data Manager II
Clinical Data Manager specialising in EDC systems, CDISC standards, and GCP-compliant data governance. Creator of the Clinical Research Learning Hub — a platform built to make rigorous clinical research education accessible to every professional in the field.
Connect on LinkedInAll content is expert-written and SME-reviewed. Regulatory references are verified against current ICH GCP E6(R2), FDA, and EMA guidance.


Comments
Leave a comment