Every clinical trial follows a sequence. But most breakdowns stop at the phase level: Planning, Start-Up, Conduct, Close-Out — without showing the operational detail that actually drives execution. This guide walks through all 20 activities of a clinical study lifecycle, covering what happens, who owns it, and why each step matters from a data and operations perspective.
All activities described operate within the framework of ICH GCP E6(R2), the international standard for clinical trial conduct, with the updated ICH GCP E6(R3) now in adoption across regulatory agencies globally. Where US-specific or EU-specific requirements differ, both are noted.
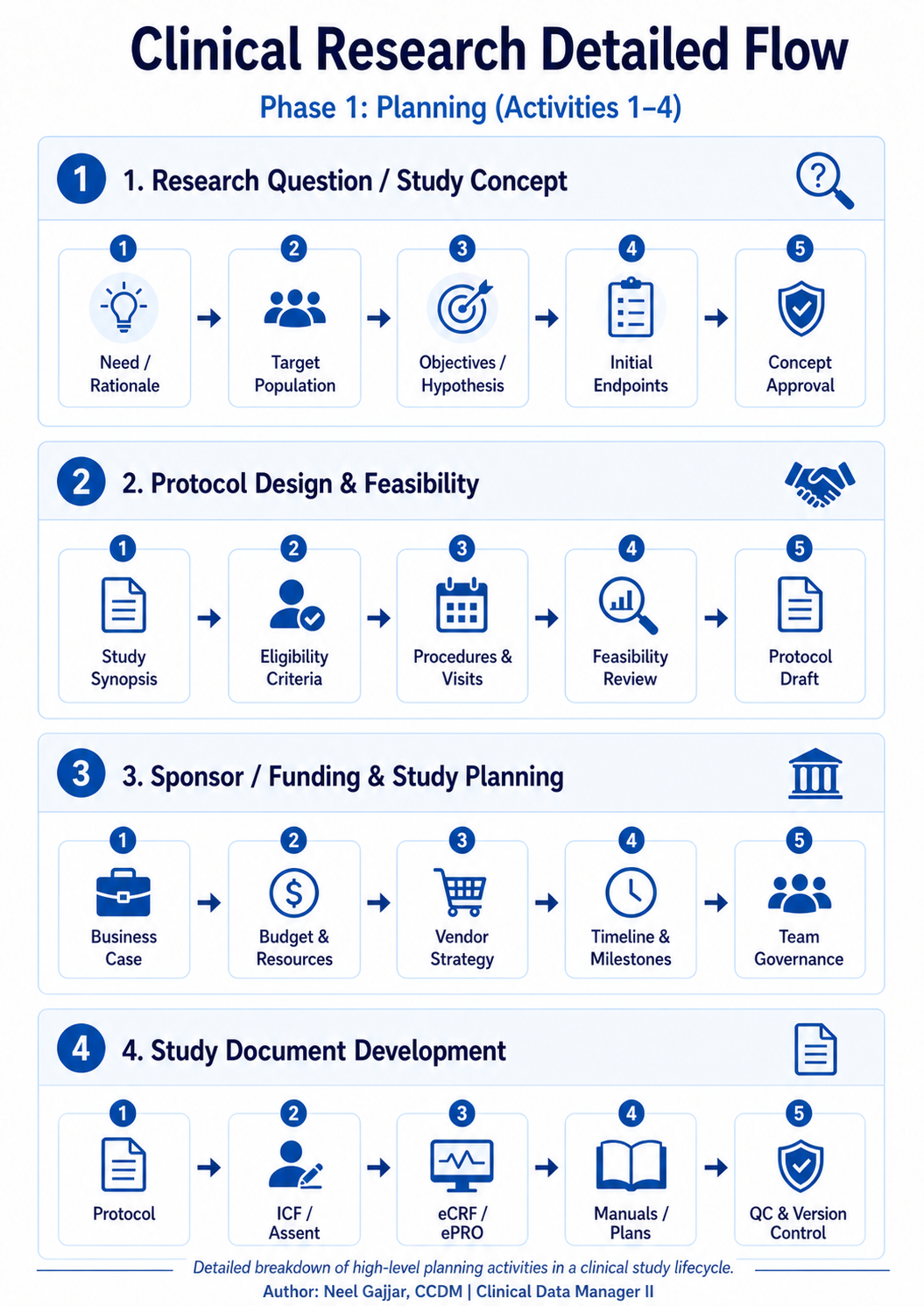
Phase 1: Planning (Activities 1–4)
The Planning phase defines the scientific and operational blueprint for the entire trial. Decisions made here on endpoints, eligibility, and document governance constrain every phase that follows. Errors introduced in Planning are the most expensive to fix.
1. Research Question & Study Concept
Every trial begins with a clearly defined scientific question. Before a single protocol is written, the team must establish the need and rationale: what gap in knowledge or unmet clinical need does this study address? The PICO framework (Population, Intervention, Comparison, Outcome) provides the structural backbone: define each element precisely, and the research question nearly writes itself.
From there, the target population is defined: who will participate, and why are they the right group to answer the question? This stage produces the initial objectives and hypotheses (primary, secondary, exploratory) and establishes the initial endpoints. The stage closes with concept approval, typically from a scientific review board or sponsor steering committee.
Getting this right is foundational. Vague endpoints at this stage cascade into protocol ambiguity, data collection gaps, and regulatory questions that are far more costly to resolve later.
2. Protocol Design & Feasibility
With the concept approved, the team moves into protocol design. This starts with a study synopsis, a condensed overview of the design that can be circulated for early feedback before the full protocol is drafted.
Eligibility criteria (inclusion and exclusion) are defined here and must strike a deliberate balance: too restrictive and recruitment becomes impossible; too broad and the data loses scientific validity. For Phase 2 and Phase 3 trials, the Estimand framework (ICH E9(R1), 2019) must be applied at this stage, precisely defining what the trial is designed to estimate and ensuring that the primary endpoint, analysis method, and intercurrent event handling are all aligned before a single patient is enrolled.
The phase closes with a protocol draft ready for sponsor, regulatory, and ethics review.
3. Sponsor / Funding & Study Planning
Parallel to protocol development, the operational framework is being built. The business case is formalized with cost projections, timelines, and scientific value proposition. For industry-sponsored trials, this includes commercial considerations; for investigator-initiated and government-funded studies, the focus is on feasibility within grant constraints and institutional resources.
Budget and resource planning determines headcount, CRO involvement, technology platforms, and lab vendors. Under ICH GCP E6(R2), sponsors cannot transfer accountability when a CRO is engaged; the sponsor remains fully responsible for oversight of all delegated activities. Timeline and milestones are locked into the project plan, and team governance structures are established.
4. Study Document Development
This activity is often underestimated and is one of the most consequential in the entire lifecycle. The full protocol is finalized. Informed Consent Forms (ICF) and assent documents are drafted for every site country and population subgroup.
eCRF and ePRO forms are designed based on the protocol visit schedule; every data point must align with CDISC CDASH (Clinical Data Acquisition Standards Harmonization) standards, which ensure downstream compatibility with SDTM and enable regulatory submissions to FDA and EMA without manual re-mapping.
Manuals and plans are developed: the Data Management Plan, Statistical Analysis Plan, Monitoring Plan, Pharmacovigilance Plan and, critically, the Risk-Based Quality Management (RBQM) Plan, now required under ICH GCP E6(R2). The RBQM Plan identifies which risks are critical to data quality and subject safety, and defines how each will be detected, assessed, and mitigated throughout the study.
Phase 2: Start-Up (Activities 5–8)
Start-Up converts the blueprint into an operational reality. It is the phase most responsible for First Patient In (FPI) delays and the phase where proactive planning pays off most visibly.
5. Regulatory & Ethics Submission
The submission package protocol, ICF, investigator brochure, and supporting documents are compiled and submitted for regulatory and ethical review. In the US, an Investigational New Drug (IND) application is submitted to the FDA with a 30-day review clock. In the EU, a Clinical Trial Application (CTA) is submitted under EU CTR No 536/2014 to the national competent authority and ethics committee simultaneously.
Once approval and clearance are received, essential documents are filed in the Trial Master File (TMF). Global studies managing submissions across 20+ countries must track each approval independently; no site may begin until its own country-specific approval is in place.
6. Site Selection, Budget & Contracts
Sites are identified through feasibility data, therapeutic network knowledge, and historical performance metrics. A capability review assesses each site's infrastructure, patient access, staff experience, and current study load.
Budget negotiation covers per-patient costs, screen failure rates, and pass-through expenses. CTA (Clinical Trial Agreement) execution is the legal gate sites cannot initiate until contracts are fully executed. Contract negotiations that extend beyond 90 days erode site enthusiasm and delay patient access to potentially beneficial interventions.
7. Database / EDC Build & UAT
CRF specifications designed to CDISC CDASH standards are translated into the EDC system. All EDC systems used in regulated trials must comply with 21 CFR Part 11 (US) and EU Annex 11 (Europe), governing electronic records, electronic signatures, and audit trail requirements. Compliance is not optional it is a prerequisite for regulatory data acceptance.
Edit checks and rules are programmed, flagging missing, out-of-range, or inconsistent data at the point of entry. User Acceptance Testing (UAT) is conducted with data managers, clinical operations, and biostatistics. A poorly tested database generates noise queries on clean data, missing validation for critical fields, and post-lock amendments that require regulatory notification.
8. Site Initiation & Training
Training materials are finalized. The Site Initiation Visit (SIV) conducted in person or remotely (now widely accepted by regulators and standard in hybrid/decentralized trials) is the formal kick-off where the monitor confirms site readiness. ICH GCP E6(R2) Section 4.1.3 requires that all individuals involved in the trial are qualified by education, training, and experience. Training records are a primary target in regulatory inspections.
Phase 3: Conduct Part 1 (Activities 9–12)
Conduct is where the science meets the reality of human participants. The quality of every decision here determines the integrity of the data that will be used to make regulatory and clinical decisions.
9. Recruitment & Screening
A recruitment plan is executed through referral networks, patient registries, advertising where permitted, and community partnerships. Since 2024, the FDA requires sponsors of Phase 3 trials to submit a Diversity Action Plan with their IND, a written commitment to enrolling a representative trial population with specific strategies for recruiting underrepresented groups. This is no longer aspirational; it is a regulatory expectation.
The screening visit evaluates eligibility against protocol criteria. A formal screening decision is documented; participants proceed to enrollment or are recorded as screen failures with the reason coded. Screen failure rates exceeding 50% are common in certain indications and must be built into enrollment projections from the outset.
10. Informed Consent
Informed consent is not a form; it is a process. Risks and benefits are reviewed honestly. Understanding is verified, not assumed, before consent is obtained. Consent is only valid when signed before any study-specific procedures begin. Retrospective consent is a critical GCP violation and a persistent inspection finding.
Re-consent is required whenever the protocol is amended in a way that affects participant risk or when significant new safety information emerges. eConsent (electronic informed consent) is now widely used, with guidance from both FDA and EMA — it improves comprehension, enables remote consent, and creates a more robust audit trail than paper.
11. Enrollment & Randomization
Confirmed eligible, consented participants are enrolled and assigned a unique Subject ID. Randomization is managed through an IWRS or IVRS (Interactive Web/Voice Response System), the technology platform that assigns treatment arms, manages investigational product allocation, and generates the randomization audit trail. Breaches in blinding or allocation errors require immediate escalation and may require regulatory notification.
12. Study Intervention & Visit Conduct
Protocol assessments labs, vitals, questionnaires, and imaging are conducted in the specified sequence. Compliance and accountability for investigational product is fully documented. Protocol deviations must be distinguished from protocol violations: a deviation is any unplanned departure; a violation is a deviation that affects subject safety or data integrity and may require regulatory reporting. Both must be documented and reported to the sponsor.
Phase 4: Conduct Part 2 (Activities 13–15)
Data collected during Conduct is only as good as the processes used to capture, review, and protect it. This phase is where data management earns its place at the centre of clinical operations.
13. Data Collection
All data must adhere to ALCOA+ principles: Attributable, Legible, Contemporaneous, Original, Accurate plus the extended attributes Complete, Consistent, Enduring, and Available. Each attribute is individually auditable and collectively defines what constitutes acceptable clinical data under GCP.
Data collection is governed by a Risk-Based Monitoring (RBM) strategy per ICH GCP E6(R2). Rather than 100% Source Data Verification (SDV), modern monitoring uses centralised data review and risk signals to direct monitoring resources where they are most needed. Source Data Review (SDR) has substantially replaced blanket SDV in most industry-sponsored trials. Data quality is not an end-of-study activity continuous review throughout conduct is what makes database lock manageable.
14. Data Review & Query Management
Data review operates at two levels. System-generated queries fire automatically from edit check rules. Manual queries are raised by the DM team, medical reviewers, or CRAs following central or on-site review addressing issues that require clinical judgement. Medical coding is a parallel essential activity: Adverse Events are coded to MedDRA and concomitant medications to WHODrug. Coding accuracy directly affects safety signal detection and regulatory submission quality. Query aging, the proportion of queries open beyond 30 days, is tracked as a key data quality KPI.
15. Safety Reporting & Monitoring
SUSAR (Suspected Unexpected Serious Adverse Reaction) reporting timelines are precise regulatory requirements: fatal or life-threatening SUSARs must be reported within 7 calendar days; all other SUSARs within 15 calendar days. The CIOMS I form is the standard reporting format.
The Data Safety Monitoring Board (DSMB) provides independent, unblinded safety oversight, reviewing accumulating data at pre-specified intervals and able to recommend protocol modifications or early termination. A delayed SUSAR report is one of the most serious GCP violations and will trigger regulatory authority inspection.
Phase 5: Close-Out & Reporting (Activities 16–20)
Close-Out converts the completed trial into a permanent regulatory and scientific record. The quality of close-out work determines what can be reconstructed, defended, and published sometimes years later during an inspection.
16. Last Patient Last Visit (LPLV)
LPLV is a regulatory milestone that must be reported to health authorities and triggers the formal close-out phase across all sites. AE follow-up continues beyond LPLV until all events are resolved or stabilized. In oncology and survival studies, LPLV is distinct from Last Patient Last Contact (LPLC); survival follow-up continues until the primary endpoint event is reached or the data cut-off date is hit.
Post-study access to investigational medicinal product for participants who demonstrated benefit is both an ethical obligation and, increasingly, a regulatory requirement that must be addressed in the protocol and CSR.
17. Site Close-Out & Reconciliation
The Trial Master File (TMF), the sponsor's definitive document repository and the Investigator Site File (ISF), the site's equivalent, are reviewed for completeness. These are separate files held by different parties; both must be complete and able to stand alone during an inspection. Sponsors typically archive for 15 years post-Marketing Authorization. TMF gaps discovered after closure cannot be remediated without a formal reconstruction process; invest in TMF management throughout the study, not at the end.
18. Database Lock
Database lock happens in two distinct steps. Soft Lock: All queries resolved, medical coding finalized, final sign-offs obtained. The Data Management team retains access for CDISC conversion, transforming raw clinical data into SDTM (Study Data Tabulation Model) and ADaM (Analysis Data Model) formats required for regulatory submission.
Hard Lock: The database is completely frozen. No further changes are possible without a formal amendment with full regulatory justification. For blinded studies, hard lock immediately precedes the formal unblinding event. Lock readiness reviews should begin weeks before the target date.
19. Statistical Analysis
CDISC SDTM and ADaM datasets prepared during the soft lock window serve as the basis for all statistical outputs, required by both FDA and EMA. Tables, Listings, and Figures (TLFs) are generated. QC is performed through independent programming: a second statistician replicates all analyses independently. For registration-track studies, Integrated Summaries of Efficacy (ISE) and Integrated Summary of Safety (ISS) are also required, aggregating evidence across the full development programme.
20. Clinical Study Report & Submission
The Clinical Study Report (CSR) is structured according to ICH E3, the international guideline governing CSR content and format for regulatory submissions. Results disclosure is a legal obligation: FDA requires results to be posted to ClinicalTrials.gov within 1 year of the primary completion date under FDAAA 801; the EU requires equivalent posting to the EU Clinical Trials Register. Non-compliance carries financial penalties. All study records are archived, typically 15 years for sponsor records.
The CSR is the end product of every decision made across all 20 activities. Its quality directly and irrevocably reflects the quality of the data and operations that preceded it.
Key Takeaway
The clinical trial lifecycle is not a linear checklist; it is a complex, interdependent system where the quality of work in each activity directly determines the options available in every activity that follows. Understanding the full sequence, not just your own lane, is what separates a competent clinical researcher from an exceptional one.
These 20 activities represent the operational backbone of every regulated clinical study, regardless of phase, indication, or geography. Master them, and you understand how clinical evidence is built.
About the Author
Neel Gajjar
CCDM®Clinical Data Manager II
Clinical Data Manager specialising in EDC systems, CDISC standards, and GCP-compliant data governance. Creator of the Clinical Research Learning Hub — a platform built to make rigorous clinical research education accessible to every professional in the field.
Connect on LinkedInAll content is expert-written and SME-reviewed. Regulatory references are verified against current ICH GCP E6(R2), FDA, and EMA guidance.



Comments
Leave a comment